MolCompass ebnet den Weg zu Arzneimittelforschung ohne Tierversuche
Um dieses Problem näher zu untersuchen konzentrierte sich Sergey Sosnin, Senior Scientist in der Forschungsgruppe für Pharmakoinformatik an der Universität Wien, auf die binäre Klassifikation. Hierbei liefert ein maschinelles Lernmodell eine Wahrscheinlichkeit von 0 % bis 100 %, die angibt, ob eine chemische Verbindung aktiv ist oder nicht (z. B. toxisch oder nicht toxisch, bioakkumulierbar oder nicht bioakkumulierbar, ein Binder oder Nicht-Binder an ein spezifisches menschliches Protein). Diese Wahrscheinlichkeit spiegelt das Vertrauen des Modells in seine Vorhersage wider. Idealerweise sollte das Modell nur bei korrekten Vorhersagen Werte nahe 0% (sicher inaktiv) oder 100% (Sicher aktiv) geben. Wenn das Modell unsicher ist und eine Vertrauensbewertung von z.B. 51 % abgibt, sollten diese Vorhersagen verworfen und alternative Methoden zur Risikobewertung herangezogen werden. Ein Problem entsteht jedoch dann, wenn das Modell falsche Vorhersagen mit hohen Wahrscheinlichkeiten liefert.
„Dies ist das wahre Albtraumszenario für Toxikologen“, sagt Sergey Sosnin. „Wenn ein Modell vorhersagt, dass eine Verbindung mit 99 % Sicherheit nicht toxisch ist, die Verbindung aber tatsächlich toxisch ist, gibt es keine Möglichkeit zu wissen, dass etwas falsch gelaufen ist.“
Die einzige Lösung besteht darin, jene Bereiche des chemischen Raums – also mögliche Klassen organischer Verbindungen – im Voraus zu identifizieren, in denen das Modell „blinde Flecken“ hat, und diese zu vermeiden. Dazu müssen Forschenden, die das Modell bewerten, die vorhergesagten Ergebnisse für Tausende von chemischen Verbindungen einzeln überprüfen – eine mühsame und fehleranfällige Aufgabe.
Überwindung dieses bedeutenden Hindernisses
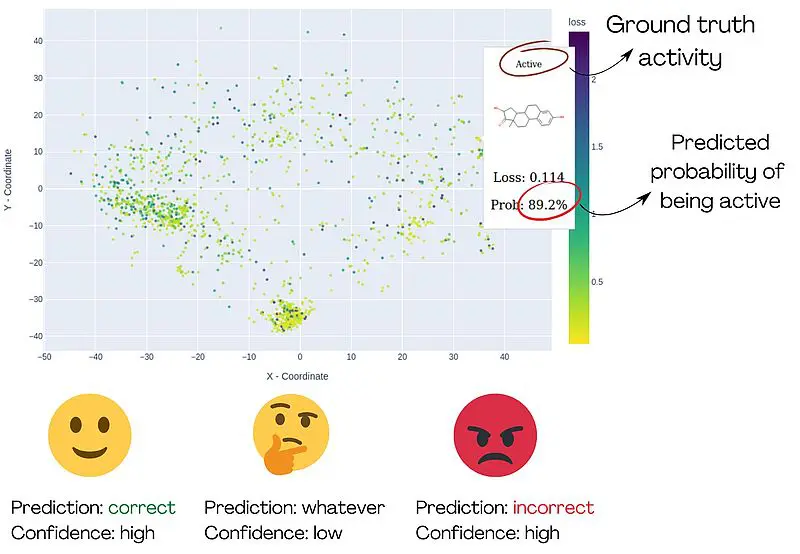
„Um diese Forschenden zu unterstützen“, fährt Sosnin fort, „entwickelten wir interaktive grafische Werkzeuge, die chemische Verbindungen auf eine 2D-Ebene projizieren, ähnlich wie geografische Karten. Mit Farben heben wir die Verbindungen hervor, die mit hoher Sicherheit falsch vorhergesagt wurden, sodass Benutzer*innen sie als Cluster roter Punkte identifizieren können. Die Karte ist interaktiv und ermöglicht es den Benutzer*innen, den chemischen Raum zu untersuchen und besorgniserregende Bereiche zu erkunden.“
Die Methodik wurde anhand eines Modells zur Bindung an den Östrogenrezeptor getestet. Nach der visuellen Analyse des chemischen Raums wurde klar, dass das Modell gut für z. B. Steroide und polychlorierte Biphenyle funktioniert, aber bei kleinen, nicht zyklischen Verbindungen völlig versagt und daher nicht für diese verwendet werden sollte.
Die in diesem Projekt entwickelte Software ist der wissenschaftlichen Community frei zugänglich auf GitHub verfügbar. Sergey Sosnin hofft, dass MolCompass Chemikern und Toxikologen zu einem besseren Verständnis der Einschränkungen von Computermodellen verhelfen wird. Diese Studie ist ein Schritt in Richtung einer Zukunft, in der Tierversuche nicht mehr notwendig sein werden und der einzige Arbeitsplatz für Toxikolog*innen ein Schreibtisch mit einem Rechner ist.
Original Paper:
Lesen Sie dazu auch:
- DGKL: Neues Verfahren macht Tierversuche überflüssig
- DGKL: VORGESTELLT: Maike Frye und die drei R
- DGKL: VORGESTELLT: Der Tumor-Hacker
Die Beiträge im News-Bereich werden erstellt vom
X-Press Journalistenbüro GbR
Schwimmbadstr. 29
37520 Osterode am Harz
Web: www.xpress-journalisten.com
E-Mail: redaktion(at)med-lab-portal.de
Gender-Hinweis. Die in diesem Text verwendeten Personenbezeichnungen beziehen sich immer gleichermaßen auf weibliche, männliche und diverse Personen. Auf eine Doppel/Dreifachnennung und gegenderte Bezeichnungen wird zugunsten einer besseren Lesbarkeit verzichtet
Meistgelesen
 Hantavirus aktuell: Nachweis, Symptome, Therapie Was ist das Hantavirus? Hantaviren sind eine Gruppe von Viren, die weltweit vorkommen und vor allem...
Hantavirus aktuell: Nachweis, Symptome, Therapie Was ist das Hantavirus? Hantaviren sind eine Gruppe von Viren, die weltweit vorkommen und vor allem...- Hantavirus: Fakten statt Panik Meldungen über drei Todesfälle im Zusammenhang mit einem Hantavirus-Ausbruch auf dem Kreuzfahrtschif...
- Deutscher Ärztetag fordert Verbot medizinischer Diagnostikangebote in Drogeriemärkten Der 130. Deutsche Ärztetag 2026 fordert den Gesetzgeber auf, medizinische Diagnostikangebote in Drog...