Tübinger Forschende entdecken Schwachstelle bei kindlichem Hirntumor
Wissenschaftler des Hertie-Instituts für klinische Hirnforschung am Universitätsklinikum Tübingen haben eine potenzielle Schwachstelle in einer Untergruppe des Medulloblastoms identifiziert, eines der häufigsten bösartigen Hirntumore bei Kindern und Jugendlichen. Die Entdeckung könnte zu schonenderen Therapien führen, die Tumore gezielt angreifen, ohne gesundes Gewebe zu schädigen. Das Projekt wurde von der Wilhelm Sander-Stiftung mit 111.000 Euro gefördert.
Hirntumore zählen nach Leukämien zu den zweithäufigsten Krebserkrankungen im Kindesalter. Das aggressive Medulloblastom entsteht im Kleinhirnbereich, der für die Koordination von Bewegungen zuständig ist. Molekulargenetische Analysen haben ergeben, dass es sich um vier unterschiedliche Erkrankungsformen handelt. In einer Untergruppe ist der Sonic-Hedgehog-Signalweg (SHH) übermäßig aktiv. Bei Erwachsenen können SHH-Inhibitoren eingesetzt werden, bei Kindern sind sie jedoch aufgrund starker systemischer Nebenwirkungen ungeeignet, da der Signalweg in der kindlichen Entwicklung eine zentrale Rolle spielt.
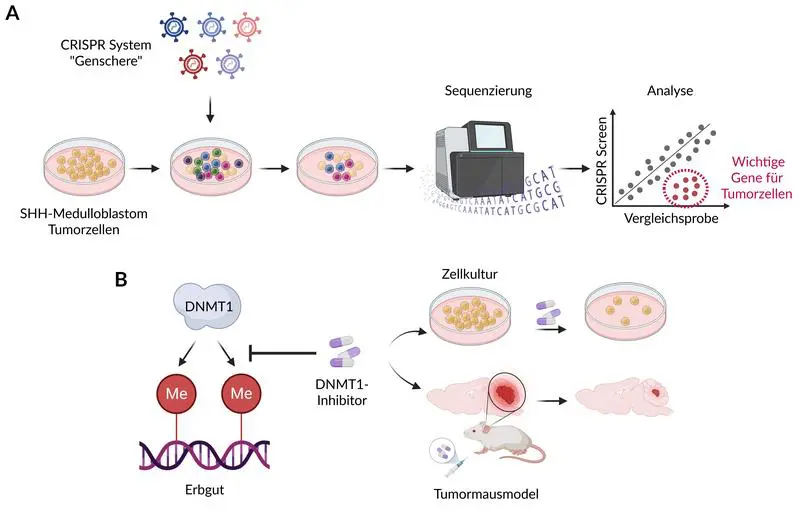
Unter Leitung von Daniel Merk nutzten die Forscher die CRISPR-Technik, um Gene in Tumorzellen systematisch auszuschalten und essenzielle Faktoren für das Tumorwachstum zu ermitteln. Die Ergebnisse zeigten, dass SHH-Medulloblastome stark von epigenetischen Regulatoren abhängen, die die Verpackung des Erbguts steuern und damit das Verhalten der Tumorzellen beeinflussen. Besonders das Gen DNMT1 erwies sich als entscheidend: Hohe Aktivität dieses Gens korreliert mit einer schlechteren Prognose bei Patienten. Obwohl DNMT1 nicht direkt zum SHH-Signalweg gehört, stört seine Abschaltung den Weg und hemmt so das Tumorwachstum.
In Tiermodellen mit genetisch veränderten Mäusen, die SHH-Medulloblastome entwickeln, reduzierten sowohl SHH- als auch DNMT1-Inhibitoren das Tumorwachstum. Eine Kombination beider Wirkstoffe erzielte den stärksten Effekt, was auch in Kulturen humaner Tumorzellen bestätigt wurde. DNMT1-Inhibitoren werden bereits bei kindlichen Leukämien klinisch angewendet und verursachen im Gegensatz zu SHH-Inhibitoren keine schweren Nebenwirkungen bei jungen Patienten.
Die Forscher sehen in ihren Ergebnissen einen vielversprechenden Ansatz für gezielte Therapien bei betroffenen Kindern. Auch für Erwachsene könnte eine Kombinationstherapie in Zukunft Vorteile bieten. Aktuell heilen 70 bis 80 Prozent der Medulloblastom-Patienten durch konventionelle Behandlungen, viele leiden jedoch unter langfristigen Folgen aggressiver Therapien.
Original Paper:
Tsiami, F., Lago, C., Pozza, N. et al. Genome-wide CRISPR-Cas9 knockout screens identify DNMT1 as a druggable dependency in sonic hedgehog medulloblastoma. Acta Neuropathologica Communincations 12, 125 (2024). doi.org/10.1186/s40478-024-01831-x
Meistgelesen
 Wundersames Fitness Probiotikum: Roseburia inulinivorans verbessert Muskelkraft massiv Forschende der Universität Granada, der Universität Almería und der Leiden University Medical Center...
Wundersames Fitness Probiotikum: Roseburia inulinivorans verbessert Muskelkraft massiv Forschende der Universität Granada, der Universität Almería und der Leiden University Medical Center...- Der persönliche Gesundheits-Check des UMG-Labors: Die Detektive des Blutes Der Persönliche Gesundheits-Check des UMG-Labors (Interdisziplinäres Labor der Universitätsmedizin G...
- NACHGEFRAGT: „Die neue GOÄ ist eine weit größere Gefahr, als das manchem klar ist“ Die neue Gebührenordnung für Ärzte (GOÄ) sieht deutliche Kürzungen für die Labormedizin vor. DGKL-Vo...